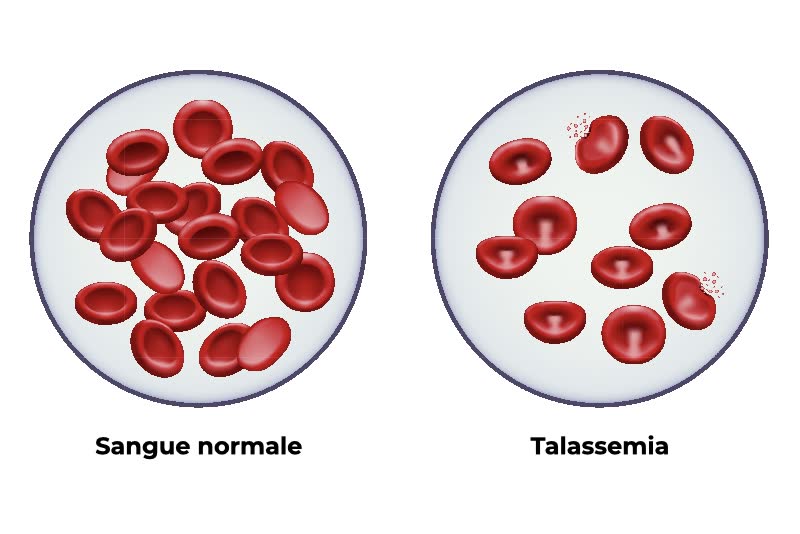
Talassemia
La talassemia è una malattia del sangue che influisce sulla produzione di emoglobina, una sostanza presente nei globuli rossi che aiuta a trasportare l’ossigeno in tutto il corpo. In persone con talassemia, il corpo ha difficoltà a produrre abbastanza emoglobina, il che può causare problemi come anemia e stanchezza.
È come se i lavoratori di una fabbrica (i globuli rossi) non avessero abbastanza strumenti (emoglobina) per fare il loro lavoro di trasportare l’ossigeno. La talassemia può variare da lieve a grave, e il trattamento dipende dal tipo e dalla gravità della malattia.
La talassemia è un’anemia ereditaria causata da mutazioni nei geni necessari per la sintesi dell’emoglobina. Ne esistono più tipi, note anche come sindromi talassemiche. Sono comuni nella fascia geografica che parte dal Mediterraneo (il nome talassemia viene dalla parola greca che significa “mare”), attraversa Balcani, Medio Oriente, Asia sudorientale e arriva alla Nuova Guinea. In Italia è particolarmente diffusa nelle isole, nelle zone costiere del Sud e nel delta padano.
- 1 Classificazione delle forme di talassemia
- 2 Sintomi e caratteristiche della talassemia
- 3 Come si diagnostica la talassemia
- 4 Come si cura la talassemia
- 5 Fonti
Classificazione delle forme di talassemia
L’emoglobina (Hb) è una molecola formata da una parte proteica (globina) e quattro gruppi eme, complessi nei quali è inserito un atomo di ferro in grado di legare una molecola di ossigeno. L’emoglobina è contenuta nei globuli rossi (o eritrociti) che la utilizzano per il trasporto dell’ossigeno dai polmoni ai tessuti.
La globina è costituita da quattro catene polipeptidiche (lunghe catene di amminoacidi), uguali a due a due. Il tipo di catene che compongono l’emoglobina (alfa, beta, gamma, delta, epsilon, zeta), codificate da sei diversi geni, varia durante lo sviluppo; nell’adulto, l’emoglobina contiene due catene alfa e due catene beta. Alla base delle talassemie vi sono difetti in uno o più dei geni che codificano le catene dell’emoglobina. Dal punto di vista clinico i difetti più rilevanti sono quelli che interessano i geni che codificano le catene alfa e beta.
Cos’è l’alfa-talassemia (α-talassemia)
L’α-talassemia (alfa-talassemia) è dovuta nella maggioranza dei casi alla perdita, detta delezione, di una o più copie del gene che codifica per la catena alfa, con conseguente eccesso di catene beta. Nella situazione normale un individuo ha quattro copie del gene, due per ciascuna delle due copie del cromosoma 16. Se manca una sola copia del gene, l’individuo è portatore della malattia; se mancano due copie, vi è una lieve alterazione ematologica, che non compromette lo stato di salute generale; se mancano tre copie, si ha la forma severa della malattia; l’assenza di tutte e quattro le copie è invece incompatibile con la vita.
Cos’è la beta-talassemia (β-talassemia)
La β-talassemia (beta-talassemia) dipende da mutazioni puntiformi, cioè piccole variazioni nella sequenza del gene che codifica la catena beta, che è localizzato sul cromosoma 11. Sono state identificate oltre 200 mutazioni associate alla malattia. Poiché ogni individuo ha due copie del cromosoma 11, sono possibili più combinazioni genetiche. Se la mutazione è presente in entrambe le copie del cromosoma, il paziente è detto omozigote per quella mutazione, se è presente in una sola copia è detto eterozigote. I pazienti eterozigoti hanno forme più lievi della malattia rispetto ai pazienti omozigoti. Un paziente può avere anche più mutazioni diverse. La situazione in cui il gene è completamente inattivo è indicata con la notazione β0, quella in cui il gene è parzialmente attivo con la notazione β+. Si distinguono così diverse forme di β-talassemia:
Si distinguono così diverse forme di β-talassemia:
- β-talassemia major o morbo di Cooley o anemia mediterranea (omozigoti β0/ β0 o eterozigoti composti gravi β0/ β+);
- β-talassemia intermedia (eterozigoti β0/ β+o omozigoti β/ β+);
- β-talassemia minor o tratto talassemico (eterozigoti β/ β0 o β/ β+).
Le talassemie vengono ulteriormente classificate in:
- forme trasfusioni dipendenti, in cui il paziente non può sopravvivere se non si sottopone regolarmente a emotrasfusione;
- forme non trasfusione dipendenti, in cui il paziente è in grado di sopravvivere senza trasfusioni o con trasfusioni occasionali.
Sintomi e caratteristiche della talassemia
La β-talassemia major si manifesta a partire dai 6 mesi di vita con i sintomi di:
- grave anemia;
- cute pallida;
- ittero;
- modificazioni dello scheletro, come ad esempio zigomi prominenti, ossa del cranio ispessite, e deformità delle ossa lunghe;
- ritardo di crescita;
- ingrossamento della milza (splenomegalia);
- ingrossamento del fegato (epatomegalia);
- irritabilità.
I pazienti possono avere una coagulazione del sangue anomala (ipercoagulopatia) e andare incontro ad osteoporosigià in giovane età. L’aspettativa di vita è breve: i pazienti muoiono in genere entro i 40 anni, nella maggior parte dei casi per complicanze cardiache dovute al sovraccarico di ferro. L’aspettativa di vita è tuttavia in netta crescita, grazie ai recenti sviluppi medici ed in particolare grazie alla terapia ferrochelante.
Come si diagnostica la talassemia
La talassemia può essere diagnosticata eseguendo alcuni esami del sangue, come:
- emocromo;
- esame dello striscio di sangue, nel quale si deposita una goccia di sangue su un vetrino e la si stende in modo uniforme per poi osservarla al microscopio ed individuare eventuali anomalie nell’aspetto dei globuli rossi;
- elettroforesi dell’emoglobina, nella quale si separano le catene polipeptidiche per studiare anomalie qualitative e quantitative;
- esami ematochimici, come l’analisi dei livelli di ferro, ferritina, transferrina e bilirubina.
È anche possibile ricorrere a test genetici, che permettono di individuare le mutazioni coinvolte nella malattia. Se una coppia è a conoscenza del fatto che uno o entrambi i partner sono portatori di mutazioni nei geni dell’emoglobina, si può eseguire una diagnosi prenatale e neonatale mediante amniocentesi o villocentesi durante la gravidanza.
Come si cura la talassemia
La terapia salvavita per i pazienti con talassemia trasfusione dipendente è la terapia trasfusionale, che consiste nel sottoporsi a emotrasfusioni regolari per tutta la vita. Poiché le trasfusioni di sangue portano a un accumulo di ferro, il paziente deve assumere una terapia ferrochelante a base di farmaci che legano questo metallo e permettono di eliminarlo.
Nei bambini con forme gravi di talassemia il trapianto di midollo o il trapianto di cellule staminali ematopoietiche da donatore permette di curare definitivamente la malattia. Nel 2019 l’Agenzia Europea per i Farmaci ha approvato una terapia genica (betibeglogene autotemcel) per i pazienti con forme severe di β-talassemia non β0/ β0. La terapia consiste nell’isolamento delle cellule staminali ematopoietiche del paziente, che vengono sottoposte a ingegneria genetica per inserire copie sane del gene per la catena beta. Le cellule, che a questo punto sono in grado di produrre la catena mancante, vengono poi reinfuse nel paziente.
Fonti
- Taher AT, Weatherall DJ, et al. Lancet. Thalassemia. 2018;391(10116):155-167. doi:10.1016/S0140-6736(17)31822-6
- Muncie HL, Campbell JS. Alpha and Beta Thalassemia. Am Fam Physician. 2009;80(4):339-444.
- Origa R. β-thalassemia. Genet Med. 2017;19(6):609-619. doi:10.1038/gim.2016.173
- Fondazione Theleton – Talassemie