Sclerosi laterale amiotrofica (SLA o Morbo di Lou Gehrig)
La sclerosi laterale amiotrofica, comunemente nota come SLA o malattia di Lou Gehrig, è una malattia progressiva dei motoneuroni che porta alla morte per insufficienza respiratoria. La maggior parte dei casi sono sporadici, ma ne esiste anche una forma familiare. La diagnosi può non essere semplice. Il trattamento è esclusivamente sintomatico.
- 1 Descrizione della sclerosi laterale amiotrofica (SLA)
- 2 Origini del nome ed epidemiologia della SLA
- 3 Come si presenta la sclerosi laterale amiotrofica
- 4 Come si diagnostica la sclerosi laterale amiotrofica
- 5 La SLA si può curare?
- 6 Fonti
Descrizione della sclerosi laterale amiotrofica (SLA)
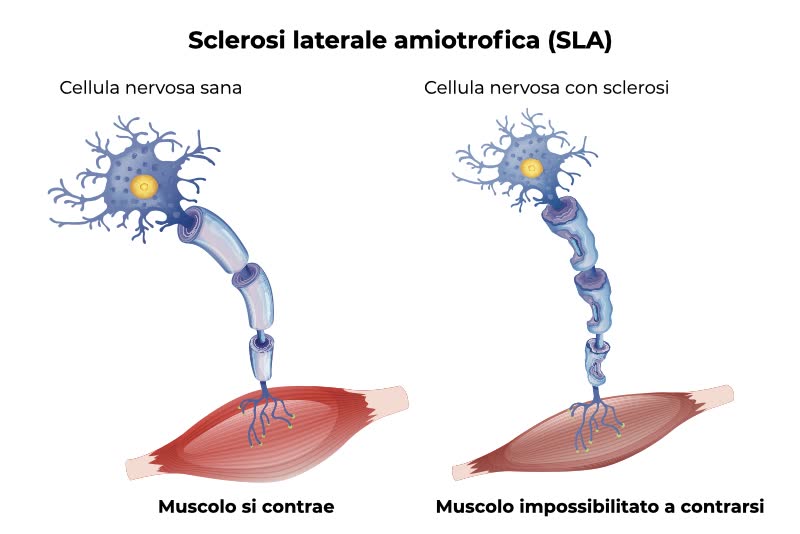
La sclerosi laterale amiotrofica (SLA) è una malattia neurodegenerativa progressiva che colpisce le cellule nervose che controllano la muscolatura (i motoneuroni) causandone la morte. La perdita dei motoneuroni porta nei pazienti alla paralisi muscolare tipica dei soggetti affetti da SLA.
Per approfondire: Differenza tra SLA e Sclerosi Multipla
Sono interessati sia i motoneuroni superiori (che partono dalla corteccia cerebrale e arrivano al tronco encefalico e al midollo spinale) sia i motoneuroni inferiori (che partono dal tronco encefalico o dal midollo spinale e raggiungono i muscoli). La malattia progredisce inesorabilmente fino a coinvolgere la maggior parte dei muscoli, inclusi i muscoli che servono alla respirazione, causando la morte del paziente per insufficienza respiratoria nell’arco di 3-5 anni. La SLA è considerata una malattia neurodegenerativa e non un disturbo neuromuscolare in quanto si è osservato che molti pazienti vanno incontro a disturbi cognitivi e/o comportamentali e che una minoranza di pazienti con SLA presenta anche demenza frontotemporale, segno di un coinvolgimento del sistema nervoso centrale.
Origini del nome ed epidemiologia della SLA
Il nome della malattia “sclerosi laterale amiotrofica” si riferisce al fatto che la degenerazione delle fibre nervose porta alla formazione di tessuto cicatriziale (sclerosi) nelle aree laterali del midollo spinale e che la perdita dei motoneuroni causa una riduzione di volume e la degenerazione funzionale dei muscoli (amiotrofia). La SLA è nota anche come “malattia di Lou Gehrig” dal nome di un giocatore di baseball statunitense che si ritirò dall’attività sportiva nel 1939 dopo avere ricevuto la diagnosi, facendo conoscere questa malattia all’opinione pubblica.
In Europa ogni anno si verificano 2-3 nuovi casi di SLA ogni 100.000 persone. Circa il 90% dei casi sono sporadici (non vi sono elementi che facciano sospettare una predisposizione genetica). Colpisce prevalentemente gli individui di sesso maschile (il rapporto maschi:femmine è di circa 2:1) e solitamente si manifesta attorno ai 60 anni. Il 10% dei casi sono ereditari; la forma familiare colpisce in ugual misura i due sessi e può insorgere anche durante la tarda adolescenza e l’inizio dell’età adulta. Sono stati identificati almeno 30 geni collegati alla SLA.
Per approfondire: Il mio inizio con la SLA: come affrontare la diagnosi?
Tra i calciatori e i giocatori di football americano è stata osservata un’incidenza di SLA più elevata di quella riscontrata nella popolazione generale, tuttavia gli studi sull’associazione tra sport e SLA non hanno dato risultati conclusivi. Allo stesso modo non ci sono dati certi riguardo ad altri fattori che sono stati proposti come fattori di rischio, tra cui l’esposizione ad alcune tossine o alle radiazioni elettromagnetiche, ad eccezione dei dati che collegano il fumo e i casi sporadici di SLA che sembrano convincenti.
Come si presenta la sclerosi laterale amiotrofica
La sclerosi laterale amiotrofica si presenta in modo eterogeneo. La prima manifestazione della SLA in due terzi dei pazienti è la debolezza dei muscoli degli arti dovuta alla perdita dei neuroni spinali (SLA a esordio spinale). Gli altri sintomi includono la difficoltà nell’articolare le parole, nel masticazione e nella deglutizione dovuta alla perdita dei motoneuroni della regione bulbare del sistema nervoso, localizzata tra il cervello e il midollo spinale (SLA a esordio bulbare).
Per approfondire: La SLA e i sintomi da non sottovalutare
Altri sintomi della SLA sono rigidità (spasticità) e contrazioni muscolari involontarie (fascicolazioni), talvolta dolorose (crampi muscolari). La compromissione dei muscoli respiratori fa sì che la tosse sia poco efficace e che il respiro diventi debole. I neuroni che innervano i muscoli dell’occhio e degli sfinteri della vescica e dell’intestino solitamente sono risparmiati dalla malattia.
Come si diagnostica la sclerosi laterale amiotrofica
In genere tra l’insorgenza dei primi sintomi e la diagnosi passano 12 mesi. Non esiste nessun test specifico, perciò è necessario escludere altre possibili cause dei sintomi e avere riscontro della progressione della malattia. La diagnosi si basa sul quadro clinico, sull’elettromiografia e su indagini che servono ad escludere altre malattie che si presentano come la SLA. La funzione respiratoria viene valutata mediante spirometria e pletismografia.
La SLA si può curare?
Ad oggi, non esiste una cura per la sclerosi laterale amiotrofica. Sono stati identificati solo due farmaci, il riluzolo e l’edaravone, che possono rallentare moderatamente la degenerazione motoria e ritardare il ricorso alla ventilazione meccanica. Un approccio multidisciplinare volto a gestire i sintomi della malattia può però ridurre il numero e la durata dei ricoveri in ospedale, migliorare la qualità della vita dei pazienti con SLA e persino prolungarla.
Uno dei sintomi più disturbanti è l’eccessiva produzione di saliva (scialorrea), comune nei pazienti con SLA bulbare e negli ultimi stadi della malattia, che può essere controllata farmacologicamente con farmaci anticolinergici o con iniezioni di tossina botulinica nelle ghiandole salivari; per evitare l’aspirazione delle secrezioni salivari nelle vie respiratorie è importante mantenere una tosse efficace, ricorrendo a tecniche di fisioterapia respiratoria ed eventualmente a dispositivi meccanici per assistere la tosse.
Per approfondire:
Per ovviare ai problemi di deglutizione (disfagia) si apportano variazioni nella dieta (es. scegliendo cibi con diversa consistenza) e se è necessario si ricorre al sondino naso-gastrico (nutrizione enterale). Quando compaiono sintomi di insufficienza respiratoria finché è possibile si ricorre alla ventilazione meccanica non invasiva. La ventilazione invasiva tramite tracheotomia è un metodo poco diffuso, anche se efficace per trattare i disturbi di respirazione nella SLA.
Fonti
- Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162-172. doi:10.1056/NEJMra1603471
- Hardiman O, Al-Chalabi A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071. doi:10.1038/nrdp.2017.71
- Van ES MA, hardiman O, et al. Amyotrophic lateral sclerosis. 2017;390(10107):2084-2098. doi:10.1016/S0140-6736(17)31287-4
- Fondazione Italiana di Ricerca per la Sclerosi Laterale Amiotrofica (ARISLA)