Sindrome del QT corto (SQTS)
La sindrome del QT corto è una malattia cardiaca ereditaria altamente letale. Si manifesta in persone giovani e apparentemente sane. È caratterizzata da un problema di ripolarizzazione delle cellule cardiache visualizzabile con l’elettrocardiogramma.
Per ridurre il rischio di morte improvvisa solitamente si utilizza un defibrillatore cardiaco impiantabile.
Cos'è la sindrome del QT corto
La sindrome del QT corto (SQTS, dall’inglese Short QT Syndrome) è una rara malattia cardiaca ereditaria caratterizzata da aritmie ventricolari che portano alla sincope o alla morte cardiaca improvvisa. La morte cardiaca improvvisa è un evento che colpisce persone giovani e altrimenti sane: è una delle più frequenti cause di morte improvvisa nei giovani atleti. Dipende spesso da una malattia aritmogena, una patologia che coinvolge i meccanismi che controllano il ritmo cardiaco. Le malattie aritmogene congenite che sono state associate alla morte cardiaca improvvisa includono la sindrome di Brugada, la tachicardia ventricolare polimorfa catecolaminergica (CPVT), la sindrome del QT lungo (LQTS; Long QT Syndrome) e, appunto, la sindrome del QT corto.
La sindrome del QT corto è una patologia che è stata descritta solo di recente e di cui sono stati riportati pochi casi, perciò, la sua storia naturale è ancora in via di caratterizzazione.
Con il termine “intervallo QT” ci si riferisce al tracciato dell’elettrocardiogramma (questo esame cardiologico fondamentale è descritto in dettaglio nella scheda dedicata all’argomento). Il cuore funziona come una pompa in quanto è in grado di contrarsi e rilasciarsi ritmicamente in risposta a un impulso elettrico.
L’impulso parte da una struttura, il nodo senoatriale, il “pacemaker” naturale del cuore, per passare poi ad altre strutture del sistema di conduzione del cuore. Le cellule nervose e le cellule muscolari hanno una proprietà caratteristica che le distingue dagli altri tipi di cellule: l’eccitabilità, ossia la capacità di reagire a uno stimolo.
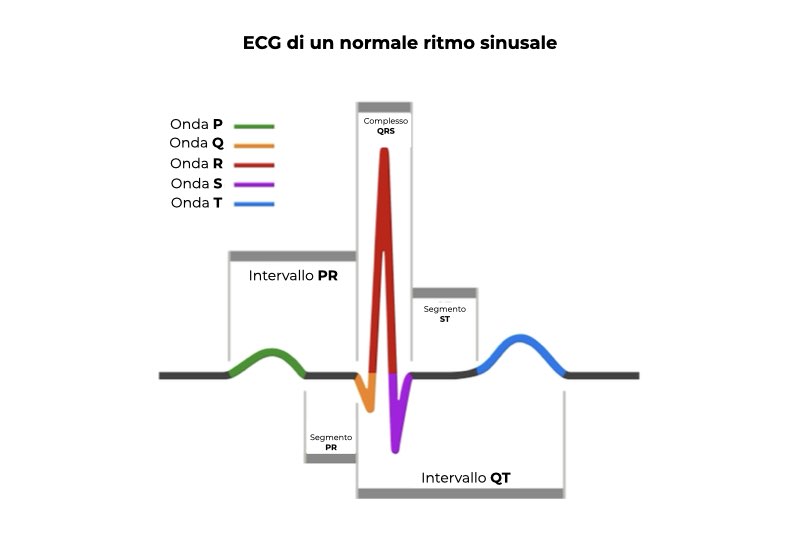
Le molecole cariche (ioni) non sono distribuite in modo omogeneo all’interno e all’esterno della cellula e questo fa sì che tra il lato interno e il lato esterno della membrana cellulare si crei una differenza di potenziale elettrico (potenziale di membrana); tutte le cellule posseggono un potenziale di membrana negativo. In risposta a uno stimolo, la permeabilità della membrana delle cellule eccitabili cambia e il repentino ingresso di molecole positive nella cellula causa una breve depolarizzazione. Questo cambiamento nel potenziale di membrana, detto potenziale d’azione, consente la trasmissione degli impulsi nervosi nei neuroni e la contrazione delle cellule muscolari. Dopo la contrazione le cellule si rilasciano (si ripolarizzano, ossia il potenziale di membrana torna al valore di riposo) per prepararsi ad un nuovo ciclo. L’elettrocardiogramma consente di visualizzare la depolarizzazione e la ripolarizzazione degli atri e dei ventricoli del cuore sotto forma di onde (onde P, Q, R, S e T).
L’intervallo QT rappresenta l’intervallo di tempo che intercorre tra l’inizio della depolarizzazione dei ventricoli e la fine della ripolarizzazione dei ventricoli. L’elemento caratteristico della sindrome del QT corto è un intervallo QT inferiore a 340 millisecondi.
Alla base della sindrome vi sono mutazioni genetiche che modificano la funzionalità dei canali per gli ioni positivi (potassio e calcio) alterando il processo di ripolarizzazione. L’età media in cui la sindrome si manifesta è attorno ai 30 anni, ma sono stati riportati casi in bambini di pochi mesi e in pazienti anziani.
Come si presenta la sindrome del QT corto
La presentazione clinica più frequente della sindrome del QT corto è l’arresto cardiaco. In alcuni casi si manifesta con palpitazioni, sincope, aritmie ventricolari e fibrillazione atriale. La prima manifestazione della sindrome è nella maggior parte dei casi l’arresto cardiaco, seguito dalla sincope. Non è stato riconosciuto nessun elemento scatenante, perciò questi eventi possono verificarsi sia durante l’attività fisica che a riposo.
Come si diagnostica la sindrome del QT corto
La diagnosi della sindrome del QT corto si basa sui sintomi, sulla storia familiare del paziente e sull’elettrocardiogramma. Il medico raccoglie informazioni sulla presentazione dei sintomi compatibili con la sindrome nel paziente e sulla presenza in famiglia di casi di sincope, morte improvvisa o fibrillazione atriale in giovane età. Il test del DNA può rivelare la presenza di specifiche mutazioni genetiche, tuttavia un test genetico negativo non esclude la diagnosi di SQTS perché non si conoscono ancora tutte le varianti genetiche coinvolte.
La probabilità che il paziente soffra di sindrome del QT corto viene calcolata usando lo Schwartz Score, un punteggio che tiene conto della durata dell’intervallo QT, dei sintomi, della storia familiare e della presenza di mutazioni genetiche note.
Come si cura la sindrome del QT corto
A causa dell’elevato rischio di morte improvvisa, ai pazienti con sindrome del QT corto viene solitamente impiantato un defibrillatore cardiaco impiantabile (ICD, Implantable Cardioverter Defibrillator), un dispositivo che rileva l’aritmia potenzialmente fatale ed eroga uno shock elettrico per riportare il ritmo cardiaco alla normalità. Nei pazienti molto giovani o quando l’impianto dell’ICD è controindicato si ricorre alla terapia farmacologica con la chinidina, un farmaco antiaritmico.
Fonti
- Dewi IP, Dharmadjati BB. Short QT syndrome: The current evidences of diagnosis and management. J Arrhythm. 2020;36(6):962-966. doi:10.1002/joa3.12439;
- Bjerregaard P. The diagnosis and management of short QT syndrome. Heart Rhythm. 2018;15(8):1261-1267. doi:10.1016/j.hrthm.2018.02.034;
- Campunzano O, Sarquella-Brugada G, et al. Recent advances in short QT syndrome. Front Cardiovasc Med. 2018;5:149. doi:10.3389/fcvm.2018.00149.